Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2024; 33(1): 1-17
Published online February 29, 2024
https://doi.org/10.5607/en23036
© The Korean Society for Brain and Neural Sciences
An Autopsy-proven Case-based Review of Autoimmune Encephalitis
Yu-Mi Shim1†, Seong-Ik Kim1†, So Dug Lim2, Kwanghoon Lee1, Eric Eunshik Kim1, Jae Kyung Won1 and Sung-Hye Park1,3*
1Department of Pathology, Seoul National University College of Medicine, Seoul 03080,
2Department of Pathology, KonKuk University School of Medicine, Seoul 05029,
3Institute of Neuroscience, Seoul National University College of Medicine, Seoul 03080, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-2072-3090, FAX: 82-2-743-5530
e-mail: shparknp@snu.ac.kr
†These authors contributed equally to this article.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Autoimmune encephalitis (AIE) is a type of immunoreactive encephalitic disorder and is recognized as the most prevalent noninfectious encephalitis. Nevertheless, the rarity of definitive AIE diagnosis through biopsy or autopsy represents a significant hurdle to understanding and managing the disease. In this article, we present the pathological findings of AIE and review the literature based on a distinct case of AIE presenting as CD8+ T-lymphocyte predominant encephalitis. We describe the clinical progression, diagnostic imaging, laboratory data, and autopsy findings of an 80-year-old deceased male patient. The patient was diagnosed with pulmonary tuberculosis 6 months before death and received appropriate medications. A week before admission to the hospital, the patient manifested symptoms such as a tendency to sleep, decreased appetite, and confusion. Although the patient temporally improved with medication including correction of hyponatremia, the patient progressed rapidly and died in 6 weeks. The brain tissue revealed lymphocytic infiltration in the gray and white matter, leptomeninges, and perivascular infiltration with a predominance of CD8+ T lymphocytes, suggesting a case of AIE. There was no detectable evidence of viral infection or underlying neoplasm. The autopsy revealed that this patient also had Alzheimer’s disease, atherosclerosis, arteriolosclerosis, and aging-related tau astrogliopathy. This report emphasizes the pivotal role of pathological examination in the diagnosis of AIE, especially when serological autoantibody testing is not available or when a patient is suspected of having multiple diseases.
Graphical Abstract
Keywords: Autoimmune diseases of the nervous system, Encephalitis, Autopsy, CD8-positive T-lymphocytes, Noninfectious disease
INTRODUCTION
Autoimmune encephalitis (AIE) is a rare, recently recognized disease characterized by inflammation in the brain parenchyma. It rapidly progresses into pronounced neurological impairment, predominantly within less than six weeks [1, 2]. AIE is considered the most common form of non-infectious encephalitis and is caused by autoantibodies that target specific neuronal epitopes, including synaptic surface structures like receptors or ion channels as well as various intracellular antigens [1, 2].
There is a degree of confusion regarding the terminology used. Historically, cell surface antigen is referred to as AIE, and intracellular antigen is associated with paraneoplastic encephalitis [3]. However, the presence of an intracellular antigen does not invariably indicate an association with a tumor, just as the existence of a cell surface antigen does not guarantee the absence of a tumor association. In this paper, the term “autoimmune encephalitis” is utilized to encompass both the traditionally used terms “paraneoplastic encephalitis” and “autoimmune encephalitis” in a comprehensive manner.
The exact cause of AIE is often unknown. In some cases, the immune system produces specific antibodies, as observed in the initial description of AIE in patients with ovarian teratomas [4]. In 1938, Brouwer and Biemond reported the first neurological neoplasia syndrome in a patient with ovarian cancer, characterized by cerebellar degeneration [5]. This syndrome was initially termed paraneoplastic encephalitis by Dalmau and colleagues in 2007, who later described cases of the N-methyl-D-aspartate receptor (NMDAR)-associated encephalitis treated with oophorectomy and immunotherapy [4]. In their study, twelve women aged 14 to 44 years presented with severe neurological symptoms linked to specific brain-targeting antibodies [4]. Most had ovarian teratomas and removal of it led to improvements of symptoms, while those untreated faced severe neurological symptoms or death. The neurological symptoms included severe psychiatric symptoms, amnesia, seizures, and other complications. Subsequently, this clinical syndrome was given the name “AIE” and encompasses the broader spectrum of encephalitis not associated with infection or neoplasia [6, 7].
Clinically, the differential diagnosis of AIE includes primary inflammatory diseases of the central nervous system (CNS), such as vasculitis or demyelinating disease, and systemic inflammatory diseases like Behçet’s disease, lupus, bacterial or viral infection including herpes simplex virus [2]. Other possibilities taken into account for differential diagnosis include metabolic, neoplastic, toxic or drug, and psychiatric disorders [8]. It is important to note that AIE may manifest with clinical, radiological, electrophysiological, and laboratory findings that resemble those of other serious brain disorders like sporadic Creutzfeldt-Jakob disease (sCJD) [9]. Due to its rarity, AIE can be misdiagnosed as psychiatric or neurological disorders or even drug abuse [10, 11]. However, AIE is considered a treatable disease, and early diagnosis is crucial to prevent severe complications [12].
Over the past two decades, the discovery of autoantibodies has rapidly expanded the field of AIE, leading to the identification of numerous new disease entities within the category of encephalitis [1, 13]. However, a significant challenge faced by clinicians is that many cases of AIE do not exhibit known autoantibodies [10]. In 2011, Najjar and colleagues proposed the term “seronegative AIE” to describe a group of immunoreactive encephalitic diseases where no identifiable neuronal autoantibodies are present in the serum and cerebrospinal fluid (CSF) [8]. The idea of seronegative AIEs initially had a debate, recently established diagnostic criteria define this subset of cases [1, 11].
AIE presents a diagnostic challenge as an inflammatory brain disease, with relatively few reported cases in the literature that have undergone brain biopsy or autopsy [4, 12]. Recently, a distinct form of CNS immune reconstitution inflammatory syndrome (IRIS) known as ‘CD8 encephalitis’ has been recognized [14, 15]. This condition is characterized by the infiltration of CD8+ T cells into the brain in the absence of a high viral burden or typical IRIS presentation. Diagnostic approaches for CD8 encephalitis include the confirmation of CD8+ lymphocytes in CSF and brain tissue. However, there are limited reports available in the literature, and specific cases identified through post-mortem examinations are scarce.
In the context of this study, an autopsy case was encountered involving a patient who exhibited a rapidly progressive clinical course of encephalitis. Notably, perivascular CD8+ T-lymphocytes were predominantly observed throughout the entire brain tissue. This article aims to provide an overview of AIE to improve understanding of the disease through an autopsy-proven case.
MATERIALS AND METHODS
The material for this study consisted of the brain of a deceased patient who had generously donated their brain to the Seoul National University Hospital (SNUH) Brain Bank. This donation was made with the informed consent of the patient's next of kin: their spouse. Prior to conducting any procedures, we thoroughly reviewed the patient's clinical information.
A brain-only autopsy was then carried out at the SNUH Brain Bank, following the standard protocols and in accordance with the Institutional Review Board (IRB) approval (IRB approval number 1808-087-966). The autopsy procedure adhered to established guidelines.
For histopathological analysis, formalin-fixed, paraffin-embedded (FFPE) blocks were prepared and used. Various staining techniques, such as hematoxylin and eosin (H&E), luxol fast blue (LFB), immunohistochemistry (IHC) were employed to examine the brain tissue.
IHC analysis was performed using specific primary antibodies in FFPE sections. The primary antibodies (Table 1) used in this study included NeuN (1:500, Millipore, Temecula, USA), synaptophysin (1:200, Novocastra, Neuwcastle, UK), GFAP (RTU, Ventana), Neurofilament (NF, 1:2000, DAKO, Glostrup, Denmark), CD3 (RTU, Ventana), CD8 (RTU, Ventana), CD20 (1:500, DAKO), CD68 (1:2000, DAKO), and TMEM119 (1:500, ABCAM, Bristol, UK), PD1, PDL-1 and stains for Luxol fast blue (LFB), myelin basic protein (MBP, 1:200, Cell Marque, Rocklin, US), aquaporin 4 (1:2000, Millipore), CMV (1:50, DAKO), and HSV (RTU, DAKO). Additional immunohistochemical stains were conducted to rule out neurodegenerative disorders, including β-amyloid (1:500, Covance, Dallas, USA), phosphorylated tau (AT8) (1:300, ThermoFisher, Waltham, USA), α-synuclein (1:1000, ABCAM), and pTDP43 (1:10000, Cosmobio, Tokyo, Japan). Positive controls for these stains were brain tissues from patients with specific neurological diseases, while a negative control was performed by omitting the primary antibodies during immunostaining. Alzheimer’s disease brain tissue was the control for pTau and beta-amyloid, Parkinson’s disease brain tissue was the control for α-synuclein, and limbic-predominant age-related TDP43 encephalopathy brain tissue was the control for TDP43.
An autopsy-proven case presentation
The deceased patient was an 80-year-old male patient who had presented with several symptoms, including decreased appetite and drowsiness one week before hospital admission. The diagnosis of pulmonary tuberculosis was made four months prior based on a chest x-ray and MTB PCR. The patient also had a medical history of diabetes and hypertension. Before the onset of initial symptoms, he was living at home for more than six months, eating regularly, using a cane for mobility, and requiring diaper assistance. At the time of the visit, the patient was taking the following medications. Ethambutol 400 mg 3t qd, isoniazid 100 mg 3t qd, rifampicin 600 mg 1t qd, pyridoxine 50 mg 1t qd, isosorbide 40 mg 1t bid, diltiazem 90 mg 1t bid, aspirin 100 mg 1t bid, evogliptin/metformin 2.5/500 mg 1t bid, atorvastatin 10 mg 1t qd, glimepride 2 mg 1t qd, pantoprazole 20 mg 1t qd.
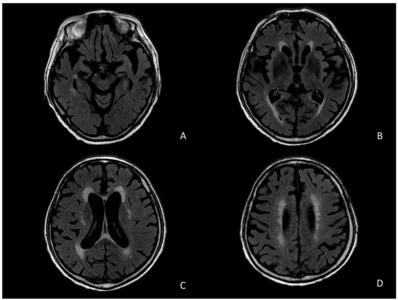
Brain magnetic resonance imaging (MRI) scans and Electroencephalogram (EEG) examination were performed to explore the cause of the mental change. The brain MRI revealed mild global atrophy of the cerebrum with chronic infarction at the right cerebellum and communicating hydrocephalus, along with prominent bilateral medial temporal atrophy (Fig. 1). MR angiography scans showed atherosclerotic irregularity of the left clinoid-ophthalmic intracerebral artery and bilateral distal vertebral artery (V4). No recent or acute lesions were observed.
EEG examination indicated generalized continuous delta slow activity, suggesting diffuse cerebral dysfunction, but no epileptiform discharge was observed.
Rapid ACTH stimulation test was performed to rule out adrenal insufficiency but showed normal cortisol and aldosterone response. Irregular peribronchial consolidation in the right upper lobe (RUL) was observed on chest CT at the time of diagnosis of tuberculosis. The RUL lesion was unremarkable on the follow-up chest X-ray performed during hospitalization, the blood culture was negative, and the culture and Gram stain of the respiratory specimen were also negative.
Laboratory tests showed mild leukocytosis, elevated erythrocyte sedimentation rate (ESR), and hyponatremia, which was determined to be a syndrome of inappropriate antidiuretic hormone secretion (SIADH) related to lung disease or a CNS lesion. The patient’s mental alertness and orientation improved temporally after receiving Tolvaptan, 7.5 mg 1T qid, a medication to treat hyponatremia. WBC count was 13.08×103/μl (normal range: 4.5~11×103/μl), absolute neutrophil count (ANC) was elevated into 9875/μl (normal range: 2500~7000 neutrophils/μl), and ESR was also elevated to 69 mm/h (normal range: ≤15 mm/h in male). Electrolyte measurements revealed hyponatremia with Na 127 millimoles/L (normal range: between 136~145 mmol/L), K (potassium) 3.8 (normal range: 3.6~5.2 mmol/L), low chloride 89 mmol/L (normal range: 96~106 mmol/L), and total CO2 27.8 (normal range: 23~30 mEq/L). Serologic tests including RPR, HbsAg, anti-HBs, HCV Ab, and HIV Ab showed no abnormalities.
For the chronic cerebral infarction seen on MRI, he maintained his aspirin regimen. During the hospitalization, the patient presented with vomiting with abdominal distension and an abdominal X-ray was performed. The patient was diagnosed with small bowel paralytic ileus. This improved with medication, metoclopramide q8h in a fasting state, and he continued tube feeding by day 8 of his hospitalization. He also complained of melena on the 6th day of hospitalization and underwent colonoscopy, which failed to find any bleeding focus and was thought to be a stool color change due to antituberculosis medication. But the stool occult blood test was positive. Therefore, lansoprazole and rebamipide was prescribed.
Initially, AIE was not suspected, so autoantibody tests had not been conducted. After a 13-day hospitalization, the patient improved and was discharged. The following medications were prescribed at the time of discharge. Ethambutol 400 mg 3t qd, isoniazid 100 mg 3t qd, rifampicin 600 mg 1t qd, pyridoxine 50 mg 1t qd, atorvastatin 10 mg 1t qd, Tolvaptan 7.5 mg 1t qd, isosorbide 40 mg 1t bid, Itopride 50 mg 1t tid, rebamipide 100 mg 1t bid, aspirin 100 mg 1t bid, diltiazem 90 mg 1t bid, lansoprazole 15 mg 1t qd. He died a month later in a nursing home due to exacerbation of pneumonia.
Autopsy findings
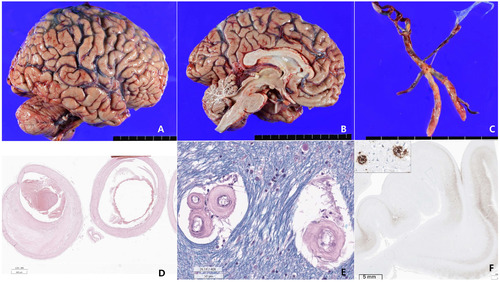
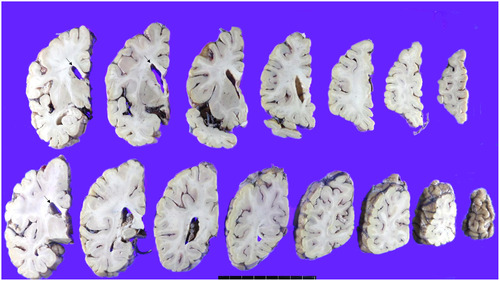
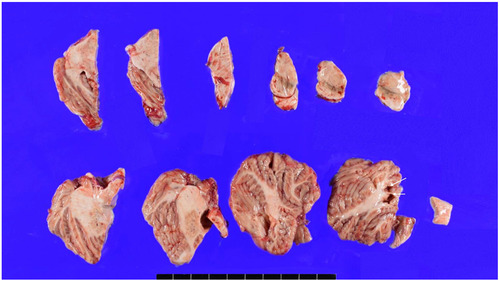
The patient’s brain weight was 1290 grams and gross examination of the brain demonstrated mild brain atrophy without focal lesions (Fig. 2A, 2B, 3, 4). Severe atherosclerosis was observed in the circle of Willis, with more than 50% occlusion of the lumen in more than one artery, corresponding to a score of 3 by Vascular cognitive impairment neuropathology guidelines (VCING) and score 3 [16] by Esiri et al’s semiquantitative scale (Fig. 2C, D) [17]. Arteriolosclerosis was markedly present in the entire brain (Fig. 2E). IHC for phosphorylated tau (AT8) IHC indicated the presence of neurofibrillary tangles and neuropil threads in the temporal lobe, classifying the case as Braak stage III out of VI (Fig. 2F). Additionally, β-amyloid IHC demonstrated the presence of classic amyloid cored plaques (shown in the inlet of Fig. 2F), presented in the whole neocortex, hippocampus, thalamus, amygdala, basal ganglia, and midbrain (Thal phase was 4/5) (Fig. 2F). These pathological features were observed throughout various brain regions, including the neocortex, hippocampus, thalamus, amygdala, basal ganglia, and midbrain, with a Thal phase of 4 out of 5. Consequently, based on the NIA-AA criteria, the patient is categorized as having an intermediate level of Alzheimer’s neuropathologic changes.
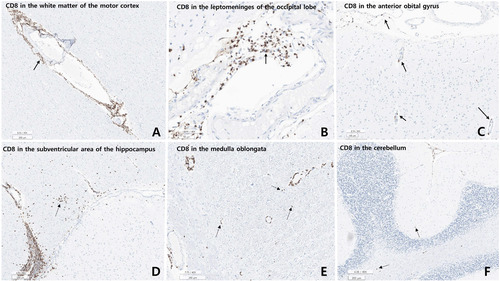
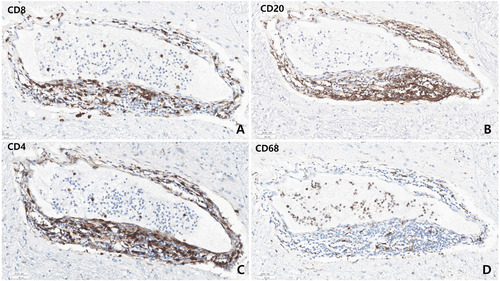
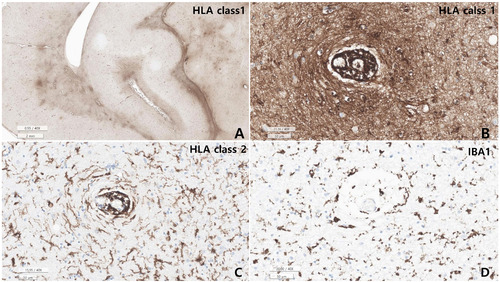
Microscopic examination revealed varying degrees of inflammation throughout the entire brain, both gray and white matter, including the basal ganglia, thalamus, brainstem, spinal cord, and meninges. The inflammatory infiltrates were primarily perivascular and extended into the parenchyma (Fig. 5, 6). There was no evidence of demyelination or loss of GFAP stain in the reactive astrocytes, indicating an absence of astrocyte damage. However, moderate transcortical gliosis and subpial gliosis were observed. CMV, HSV and adenovirus IHC were negative. Most of the inflammatory infiltrates consisted of CD8+ T-cells, suggesting AIE (Fig. 6). B lymphocytes are also found, mainly in the perivascular area and rarely in the parenchyma, but plasma cells were not identified. CD68 and IBA-1 immunostaining revealed marked microglial activation and the presence of macrophages in the cortex and white matter. The human leukocyte antigen (HLA) class I was overexpressed in the brain tissue, especially in the white matter and perivascular area, and class II was robustly positive in the microglia (Fig. 7). There were no signs of acute hypoxic-ischemic leukoencephalopathy or severe edema in the cortices. Neither vasculitis nor brain tissue necrosis was present, which ruled out primary angiitis of CNS.
Further neuropathological examination revealed Alzheimer’s disease neuropathology, characterized by the presence of neuritic plaques neurofibrillary tangles, and neuropil threads. These features were well delineated through immunostains for beta-amyloid and phosphorylated Tau (AT8). The patient’s neuropathological National Institute on Aging and the Alzheimer’s Association (NIA-AA) score of AD was an intermediate stage (NIA-AA score A3, B2, C2, Thal phase 4/5, Braak stage III/IV, CERAD score 2/3, cerebral amyloid angiopathy score 0). Vascular dementia was suggested, attributed to arteriolosclerosis and atherosclerosis of the circle of Willis (score 3). Age-related Tau astrogliopathy (ARTAG) was present as well. However, there was no evidence of a-synucleinopathy, TDP43-encephalopathy, or frontotemporal lobe degeneration.
DISCUSSION
Definition and prevalence of AIE
AIE is defined as “A form of encephalitis that occurs as a result of a brain-specific immune response with or without underlying carcinoma. It is usually associated with an antibody against a neuronal, or glial, cell surface or cytoplasmic antigen. These features are different from those of paraneoplastic encephalitis, which almost always associated with cancer, and the target antigens are usually intracellular [1].”
AIE, immune-mediated inflammatory disease primarily affecting the cerebral cortex, is characterized by non-infectious origin. The prevalence of AIE is estimated to be approximately 13.7 cases per 100,000 individuals, with an incidence rate of 0.8 cases per 100,000 persons-years [18]. AIE is a type of encephalitis in which inflammation of the brain parenchyma needs to be demonstrated [19]. Half of the confirmed cases with identified cause of encephalitis result from infectious agents, such as Japanese B encephalitis, dengue fever, or herpes virus, while the other half is attributed to autoimmune diseases. Among autoimmune subclasses, acute disseminated encephalomyelitis (ADEM) and NMDAR encephalitis are the most commonly observed. Meanwhile, the prevalence of seronegative AIE is higher than 25% of the prevalence of seropositive AIE [11, 18].
The diagnostic criteria for AIE can be dependent on the specific subtype. Nevertheless, there exists a set of guidelines that can aid in identifying potential cases of AIE [2].
According to the International Encephalitis Consortium [1, 11, 19] clinical diagnostic criteria for encephalitis and encephalopathy of presumed infectious or autoimmune etiology necessitates a changed mental state, which is characterized by a reduced level of consciousness and an altered personality, that persists for a minimum of 24 hours. Additionally, minor criteria include a fever of 38 degrees Celsius or higher within 72 hours, new-onset seizure or focal neurological deficit, and white blood cells in CSF greater than or equal to 5 cells/mm3. An MRI scan is a good modality for showing brain parenchymal abnormalities, and unique T2 hyperintensity lesions. An EEG reveals an abnormality consistent with encephalitis. It is also characterized by an autoantibody profile and antibody-specific clinical and subclinical findings.
Known autoantibodies and associated clinical features
AIE is delineated by the existence of autoantibodies that specifically target diverse neuronal intracellular, cell surfaces, or synaptic proteins [3]. Table 2 shows a compilation of predominant autoantibodies associated with AIE [20, 21]. Among these, the anti-NMDAR (N-methyl-D-aspartate receptor) antibodies are particularly prominently recognized. Individuals diagnosed with anti-NMDAR encephalitis commonly manifest psychiatric abnormalities, convulsions, motor disturbances, and autonomic irregularities [1, 22]. As the disease progresses, individuals may develop seizures, movement abnormalities such as orofacial dyskinesias and limb rigidity, speech disturbances, and a decrease in consciousness. In more severe cases, autonomic dysfunction can arise, causing instability in heart rate, blood pressure, and temperature, and some patients may even require mechanical ventilation due to hypoventilation [23]. Those with anti-LGI1 (Leucine-rich glioma-inactivated 1) antibodies may exhibit faciobrachial dystonic seizures, and cognitive impairments, among other clinical signs [24, 25]. Anti-CASPR2 (Contactin-associated protein-like 2) antibodies correlate with encephalitis, peripheral nerve excitability, and Morvan’s syndrome [26]. Though less frequent, anti-AMPA receptor (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) antibodies have been linked to limbic encephalitis and other related conditions [1]. Autoantibodies that target the GABA receptor, including GABA_A and GABA_B receptor antibodies, are predominantly related to limbic encephalitis and seizure episodes [27, 28]. Anti-DPPX (Dipeptidyl-peptidase-like protein-6) antibodies, albeit infrequent, may be linked to encephalitis and gastrointestinal manifestations [13, 29, 30]. The presence of anti-mGluR5 (metabotropic glutamate receptor 5) antibodies is indicative of Ophelia syndrome, a specific subtype of limbic encephalitis [12].
Anti-glycine receptor antibodies predominantly correlate with stiff person syndrome and PERM (Progressive Encephalomyelitis with Rigidity and Myoclonus) [12]. Anti-MOG-associated AIE is associated with ADEM, optic neuritis, transverse myelitis, and cerebral cortical encephalitis with epilepsy [31]. Anti-IgLON5 antibodies suggest a distinctive neurodegenerative condition characterized by sleep aberrations, bulbar symptoms, and ambulatory difficulties [12, 32, 33]. Although less common, Anti-D2 dopamine receptor antibodies have been identified in certain basal ganglia encephalitis [34]. It is important to note that this list is not exhaustive. Ongoing scientific research continues to uncover new antibodies and their associated syndromes. Clinical manifestations might exhibit considerable heterogeneity even within a specific antibody category, and the diagnostic procedures typically involve a combination of clinical manifestations, radiographic evaluations, cerebrospinal fluid examinations, and antibody assessments.
Generally, in patients with encephalitis, the cause can be specifically identified in about 45~75% of cases [19]; The most common cause was infection, followed by ADEM, and then AIE. AIE and paraneoplastic encephalitis are conditions that require treatment during a patient’s lifetime. However, there have been very few cases of brain autopsies or biopsies for AIE [35]. The current diagnostic criteria are based on various clinical factors, allowing for the accurate determination of the timing for pharmacological treatment and reducing the risk of missing the disease during a patient’s lifetime [2]. For this reason, the diagnostic criteria sometimes do not include autoantibody tests, as these tests can be time-consuming [2].
During the patient’s lifetime, our case was not considered a differential diagnosis for AIE, and the neurological evaluation was insufficient. Clinically, Anti-NMDAR encephalitis can sometimes present symptoms that may involve the gastrointestinal tract, and this case also showed signs of paralytic ileus, which raised suspicion. Moreover, certain factors, such as the occurrence of hyponatremia in LGI1 encephalitis (a type of the VGKC-complex encephalitis), were also considered as potential indicators. However, if this case were to be classified as AIE, the pathology work-up suggested that it may be attributed to an intracellular antigen. This was based on the high CD8/CD3 ratio [36] and the nearly negative results for anti-human IgG immunostaining in our case [36, 37]. Furthermore, close apposition of neurons to CD8+ T cells was frequently observed [35-37]. It is essential to note that this case did not have an accompanying systemic tumor, and the presence of an intracellular Ag target does not necessarily imply the existence of a concomitant systemic tumor.
In common AIE cases targeting intracellular antigens, the rates of accompanying tumors are as follows: Hu (Neuronal nuclear antigen) and Ma2 (Paraneoplastic antigen) have rates over >95%, while GAD (Glutamic acid decarboxylase 2) has a rate of 25% [2]. However, Hu/Ma2/GAD AIE typically presents clinically as limbic encephalitis. In cases where patients may appear normal, the presence of autoantibodies must be confirmed through testing [2]. However, in our case, the T2-weighted fluid attenuated inversion recovery (FLAIR) image did not show the typical features of limbic encephalitis.
Regarding anti-NMDAR, it is important to perform antibody testing in the CSF. In cases of anti-NMDAR encephalitis, approximately 14% of cases will show false negatives if only a blood sample is tested [38]. CSF testing offers higher sensitivity compared to serum. Unfortunately, in our case, CSF test was not performed.
Diagnostic pitfalls in autoimmune encephalitis
Several diagnostic pitfalls and challenges exist in the diagnosis of AIE (Table 3). In the realm of clinical diagnostics, the following issues are noteworthy. Firstly, the insufficient clinical data interfere an accurate diagnosis. Secondly, atypical cases pose distinct diagnostic challenges, as anomalous presentations may lead to delays or complications in diagnostic procedures. Additionally, symptoms that overlap with those of other disorders, particularly when these symptoms coincide with other neurological conditions, can complicate the diagnostic picture. Lastly, the heterogeneity in disease progression highlights the variation in symptoms and manifestation among individuals, further complicating the diagnosis.
There are several diagnostic pitfalls to consider. In many healthcare institutions, access to antibody testing may not be readily available, and obtaining results can be time-consuming, often taking several weeks [39]. Additionally, the absence of autoantibodies does not definitely rule out the possibility of an immune-mediated disorder. Conversely, a positive test does not always guarantee an accurate diagnosis.
Incorporating the response to immunotherapy as part of the diagnostic criteria is impractical because this information is typically not available at the time of symptom onset or early clinical evaluation. Some patients with AIE may not respond to immunotherapy or may require intensive and prolonged treatments that may not be readily available in most healthcare systems, unless a firm diagnosis has been established beforehand. Conversely, patients with other disorders may respond well to immunotherapy, such as those with primary angiitis of the CNS and primary CNS lymphoma [2, 9].
Seronegative autoimmune encephalitis
The concept of seronegative AIEs initially ignited a debate, which has recently progressed with the establishment of a diagnostic criteria [1, 11] (Table 4). The diagnostic approach for seronegative AIE may include assessing clinical manifestations, reviewing MRI findings, CSF examinations, and evaluating EEG results.
Roughly half of all AIE cases are seronegative [11, 40]. Seronegative AIE is commonly categorized into three subgroups: definite autoimmune limbic encephalitis (AILE), antibody-negative probable autoimmune encephalitis (ANPRA), and ADEM [11]. Among these, ANPRA has the poorest response to treatment [11]. It has been proposed that the underlying mechanism in ANPRA may involve T-cell mediated cytotoxicity. However, there have been only three cases associated with tumors in this context [41].
Clinically, AIE presents is characterized by rapid onset of symptoms, typically occurring within days to weeks. These symptoms often include working memory deficits (short-term memory loss), altered mental status, or psychiatric symptoms [18, 42]. In MRI, some patients show abnormalities in the brain, especially in the medial temporal lobes [27]. Analysis of CSF may show an increase in white blood cells (pleocytosis) or elevated protein concentration. First of all, careful examination to rule out alternative causes present with similar symptoms, like infections, metabolic disorders, or tumors, especially CNS lymphoma, should be ruled out [2]. Often, a response to immune therapies, like steroids, azathioprine, intravenous immunoglobulin (IVIG), or plasmapheresis, can be supportive of the diagnosis [29, 43]. In seronegative AIE, the absence of specific neural autoantibodies is a defining characteristic, but this does not rule out the possibility of AIE [1]. Autoantibodies may be present but undetectable in the serum, exist only in the CSF, or might not be identified yet.
CD8 encephalitis
HIV-associated CD8-encephalitis is a severe inflammatory disorder dominated by brain infiltration by CD8+ T-lymphocytes. It usually occurs in people with HIV, when the virus is controlled well by antiretroviral treatment. Clinically, HIV-CD8+ encephalitis presents as an acute or subacute deterioration of cerebral function, often progressing to coma and death unless immediately treated with corticosteroids [44]. A definitive diagnosis is made on brain biopsy or autopsy, where diffuse infiltration by CD8+ T lymphocytes is observed mainly in the white matter. In healthy brains, T-lymphocytes are not found in the parenchyma; they are only observed within the blood vessels [44]. Among published cases worldwide, 53 cases of HIV-CD8+ encephalitis have been reported in the literature [14]. HIV-CD8+ encephalitis is clinically and pathologically distinct from classical “HIV encephalitis, in which the pathology is immunohistochemically characterized by HIV-containing microglia, microglial nodules, microglial giant cells, and few T-cells [14]. Subacute to chronic subcortical dementia syndrome is associated. The consistent immune activation seen in HIV-CD8 encephalitis, as well as in other non-HIV-related encephalitis, might have a significant harmful effect. This is due to the secretion of chemokine and cytokines from the immune cells, which can compromise or damage the blood-brain barrier (BBB), and make it easier for T-cells across the BBB.
There is no unified neuropathogenetic explanation for HIV-CD8+ encephalitis, apart from the general understanding that it involves a variable and disproportionate cellular immune response to HIV [14]. A comprehensive analysis of 19 cases of CD8+ T encephalitis by Santana et al. [44] revealed that the typical MRI findings included bilateral signal changes in the FLAIR sequence, along with perivascular impregnations displaying linear and multisignal patterns upon contrast administration. Biopsy samples from 14 patients demonstrated microglial activation, reactive gliosis, and a predominant infiltration of CD8+ T lymphocytes. Our autopsied brain also showed these three findings but did not show microglial nodules around the neurons. The testing for HIV and COVID-19 yielded negative results.
CD8+ T cells play a crucial role as mediators of cytotoxic effector functions in various contexts, including infection, cancer, and autoimmunity. In cases of cancer and chronic viral infection, CD8+ T cells experience a gradual loss of cytokine production and cytotoxicity, termed T cell exhaustion. Conversely, in autoimmunity, autoreactive CD8+ T cells retain their ability to effectively cause damage to the body’s own tissue [45]. Despite variations in clinical outcomes depending on the specific context, CD8+ T cells are consistently exposed to antigens chronically across all conditions. These chronically stimulated CD8+ T cells share certain phenotypic features, as well as similarities in transcriptional and epigenetic programming, across different disease settings. Gaining a deeper understanding of the distinct CD8+ T cells may provide valuable insight into developing novel strategies for immune clearance of chronic viral infections and cancers, as well as mitigating the self-reactivity that leads to tissue damage in autoimmune conditions.
Interpretation of present autopsy case with HLA class I and HLA class II overexpression and hyponatremia
Our patient’s autoantibodies have not been tested in blood or CSF because there was no clinical suspicion. Nonetheless, CD8+ T-cell infiltration and overexpression of HLA class I in the entire brain and spinal cord, and HLA class II in microglia was identified as a very prominent finding in our patient’s brain, strongly suggesting AIE. HLA class I overexpression was accentuated in the CD8+ T-cell infiltrating areas.
Because brain parenchyma lacks resident T-cells, brain CD8+ T-cell infiltration is considered an autoimmune etiology of brain disease [15, 45]. Several reports have shown that it occurs in several encephalitic syndromes, including infections such as HIV CD8+ encephalitis [14, 44, 46].
The HLA system is a complex set of genes that code for cell-surface proteins vital for the immune system to recognize and differentiate between “self” and “non-self” entities [47]. These molecules play a central role in the regulation of immune responses. HLA class I antigens are found on all nucleated cells and platelets, excluding those in the CNS. Conversely, HLA class II antigens appear on antigen-presenting cells (APC) including B lymphocytes, dendritic cells, macrophages, monocytes, Langerhans cells, endothelial cells, and cells in the thymus called thymic epithelial cells [48].
In a healthy brain, HLA expression is relatively very low or absent [49]. Boegel et al. [49] observed that median HLA class I expression was the lowest in the retina, brain, and muscle. However, in the context of various neurological diseases, the expression patterns can change. In diseases like multiple sclerosis, Alzheimer’s, or Parkinson’s, there is an increase in HLA expression [50, 51]. This is often due to the activation of microglia and the recruitment of peripheral immune cells into the brain. The increased HLA expression can potentially facilitate the presentation of neural antigens to infiltrating T cells, further promoting inflammation.
Examining HLA expression changes can help us understand the underlying mechanisms of AIE. For instance, why certain individuals develop AIE after a seemingly harmless infection could be partially explained by specific HLA genotypes [24].
On the contrary, HLA Class II is overexpressed in microglia, which are the primary immune cells of the brain that can become activated in AIE [52]. Upon activation, they upregulate HLA Class II molecules. Once the HLA Class II molecules are upregulated on activated microglia, they can present neuronal antigens to helper CD4+ T cells [53]. If these T cells are autoreactive, they can facilitate further inflammation and potentially B-cell activation, leading to autoantibody production. Certain HLA genotypes might predispose individuals to specific types of AIE or determine the target antigen of the immune response [24]. For example, some HLA alleles, such as HLA-DRB1*1502 have been associated with a higher risk of developing anti-NMDA receptor encephalitis [22]. HLA genotyping could potentially help in stratifying AIE risk in individuals, especially if there is a known association between specific HLA alleles and certain AIE subtypes [54].
Although the exact role of CD8+ T lymphocytes in autoimmune pathogenesis is controversial, CD8+ T-cells are predominantly major histocompatibility complex class I-restricted lymphocytes [45]. Therefore, overexpression of HLA class I might be associated with CD8+ T-cell predominant AIE in the presented autopsy case (Fig. 7).
Understanding the role of HLA molecules in AIE might open doors to novel treatments. If HLA overexpression is contributing to disease progression, then modulating this expression could potentially be therapeutic like tumor immunotherapy [55]. While HLA overexpression in AIE can exacerbate the disease process, it also provides a window into the mechanistic underpinnings of the disease and offers potential therapeutic and diagnostic avenues [56].
Possible role of hyponatremia in AIE development
Hyponatremia is the most commonly observed electrolyte disorder in hospital settings and is a direct risk factor leading to death. The primary cause of hyponatremia is often a SIADH, followed by the use of diuretic medications [57]. Interestingly, 60% of patients with anti-LGI1-associated AIE also present hyponatremia due to SIADH [30]. In the case of this patient, SIADH was presumed to be the underlying cause of hyponatremia, and stabilization was easily achieved through the correction of the electrolyte imbalance. Notably, the brain autopsy of this patient revealed a significant infiltration of CD8+ T-lymphocytes. Hyponatremia can affect the function of the blood-brain barrier through the action of cytokines, leading to increased permeability and potentially contributing to AIE, either independently in the patient’s medical history or during treatment [57, 58]. However, brain changes associated with hyponatremia have previously been reported in demyelinating disease, which was not observed in the current autopsy case [59]. Unfortunately, in this case, we cannot prove a direct connection between the infiltration of CD8+ T-cells and hyponatremia or the presence of the anti-LGI1 autoantibody.
Obstacles in AIE treatment
AIE brings many therapeutic obstacles, including the hurdle of timely diagnosis (Table 3) [1]. Its initial manifestations can be ambiguous or might resemble other neurological or psychiatric disorders. Rapid diagnosis is pivotal to ensure immediate intervention and avoid lasting neurological harm. Varied autoantibodies can necessitate distinct treatment methodologies. However, the absence of detectable autoantibodies in some patients (seronegative) adds complexity to diagnostic and therapeutic decisions. While primary treatments such as corticosteroids, IVIG, and plasmapheresis suffice for a majority, a subset might necessitate intensive secondary interventions like rituximab or cyclophosphamide [13].
Certain patients might exhibit resistance or inadequate response to conventional treatments, demanding intensified or alternate therapeutic modalities. After an initial successful treatment phase, the recurrence of AIE poses another challenge, making its timely diagnosis and management [12]. Immunotherapies and immunosuppressants, while often effective, come with potential side effects ranging from infections to organ toxicities. Paraneoplastic forms of AIE, like those associated with anti-NMDA receptor antibodies and ovarian teratomas, require the identification and treatment of the underlying tumor [4]. There’s a need for regular monitoring to assess disease activity, antibody levels, and potential side effects of treatments [12].
Patients may have residual deficits after the acute phase, requiring prolonged rehabilitation. AIE management often necessitates collaboration between neurologists, immunologists, oncologists, psychiatrists, and other specialists, which can be logistically challenging [12, 13].
CONCLUSION
AIE covers a spectrum of disorders characterized by diverse neurological symptoms, making clinical differentiation historically challenging. It is important to recognize the limitations in diagnosing AIE, particularly the lack of aconfirmed autoantibody test. This autopsy case highlights the potential value of brain biopsy as an approach for diagnosing AIE, especially in patients with suspected AIE alongside other neurological disorders. Therefore, future studies should focus on performing biopsy or autopsy investigations through detailed examination of tissue sections to improve our understanding of AIE.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The authors sincerely acknowledge the technical assistance provided by the Division of Immunohistochemical Laboratory of the Department of Pathology and Brain Bank of Seoul National University Hospital.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The corresponding author's datasets for this study are available on reasonable request.
DECLARATION OF INTEREST
The authors have no conflict of interest relevant to the content of this article to declare.
FUNDING
This study was supported by a fund by Research of Korea Centers for Disease Control and Prevention (2023-ER1005-00) and the Korean Ministry of Health and Welfare's Program for Institution of Corpse-derived substance supply (2023-291).
AUTHOR CONTRIBUTIONS
Sung-Hye Park designed, wrote, and supervised the study. Yu-Mi Shim, Seong-Ik Kim, So Deok Lim, and Sung-Hye Park wrote the manuscript. Sung-Hye Park reviewed histology slides, diagnosed all pathology reports, and collected anonymized data for qualitative analysis. Seong-Ik Kim, Eric Eunshik Kim, Kwanghoon Lee, and Yu-Mi Shim participated in the autopsy and analyzed clinical, radiological, and pathological data.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tables
The information of the primary antibodies studied in this autopsy case
| Antibodies | Dilution | Clone | Antigen retrieval | Company |
|---|---|---|---|---|
| Aquaporin 4 | 1:2000 | Polyclonal | Ventana CC1 at 100°C | Millipore, Temecula, USA |
| CD3 | RTU | 2GV6 | Ventana CC1 at 100°C | Ventana |
| CD8 | RTU | SP57 | Ventana CC1 at 100°C | Ventana |
| CD20 | 1:500 | L26 | Ventana CC1 at 100°C | DAKO, Glostrup, Denmark |
| CD68 | 1:2000 | KP1 | Ventana CC1 at 100°C | DAKO, Glostrup, Denmark |
| CMV | 1:50 | CCH2+DDG9 | Ventana CC1 at 100°C | DAKO, Glostrup, Denmark |
| GFAP | RTU | EP672Y | Ventana CC1 at 100°C | Ventana |
| HSV | RTU | Polyclonal | Ventana CC1 at 100°C | DAKO, Glostrup, Denmark |
| IBA-1 | 1:12,000 | EPR16589 | Ventana CC1 at 100°C | ABCAM, Bristol, UK |
| NeuN | 1:500 | A60 | Ventana CC1 at 100°C | Millipore, Temecula, USA |
| NF | 1:2000 | 2F11 | Ventana CC1 at 100°C | DAKO, Glostrup, Denmark |
| Phosphorylated NF | 1:10,000 | NP-1 | Ventana CC1 at 100°C | Millipore, Temecula, USA |
| Synaptophysin | 1:200 | 27G12 | Ventana CC1 at 100°C | Novocastra, Newcastle, UK |
| α-synuclein | 1:1000 | EP1536Y | Ventana CC1 at 100°C | ABCAM, Bristol, UK |
| β-amyloid | 1:5000 | 6E10 | Ventana CC1 at 100°C | Biolegend, SanDiego, USA |
| 3 repeat (3R) tau | 1:200 | 8E6/C11 | Ventana CC1 at 100°C | Millipore, Temecula, USA |
| 4 repeat (4R) tau | 1:1000 | Polyclonal | Ventana CC1 at 100°C | Cosmobio, Tokyo, Japan |
| Phosphorylated-tau | 1:300 | AT8 | Ventana CC1 at 100°C | ThermoFisher, Waltham, USA |
| pTDP43 | 1:10,000 | pS409/410 | Ventana CC1 at 100°C | Cosmobio, Tokyo, Japan |
| TMEM119 | 1:500 | Polyclonal (c-ter) | Ventana CC1 at 100°C | ABCAM, Bristol, UK |
CMV, cytomegalovirus; GFAP, Glial fibrillary acidic protein; HSV, Herpes simplex virus; NeuN, Neuronal nuclei; NF, neurofilament; CD, Cluster of differentiation; p-Tau, phosphorylated-Tau; p-TDP43, Phosphorylated TAR DNA binding protein; RTU, ready to use.
The list of known autoantibodies and associated clinical features in autoimmune encephalitis
| Autoantibody | Associated clinical features |
|---|---|
| Anti-NMDAR IgG1 | - Encephalopathy with altered consciousness - Psychiatric symptoms like hallucinations, agitation - Seizures & Orofacial dyskinesias - Movement disorders such as choreoathetosis - Autonomic instability including hyperthermia, tachycardia - Coma and central apnea |
| Anti-LGI1 IgG4 | - Subacute amnesia and confusion suggestive of limbic encephalitis - Seizures, most notably faciobrachial dystonic seizures - Sleep disturbances - Hyponatremia due to syndrome of inappropriate antidiuretic hormone secretion (SIADH) |
| Anti-CASPR2 IgG4 | - Limbic encephalitis with memory deficits - Morvan syndrome, which presents with neuromyotonia, sleep disturbances, and autonomic symptoms - Peripheral nerve hyperexcitability or neuromyotonia alone |
| Anti-GABA(B)R IgG1 | - Limbic encephalitis presenting with short-term memory loss, seizures, and psychiatric symptoms - Some cases might exhibit ataxia or opsoclonus |
| Anti-AMPAR | - Acute to subacute onset limbic encephalitis - Amnesia, confusion, seizures, and psychiatric/behavioral symptoms |
| Anti-DPPX | - Multifocal encephalitis with myoclonus, tremors, and hyperekplexia - Diarrhea and weight loss |
| Anti-GlyR IgG1/3 | - Stiff-person syndrome like muscle rigidity and spasms - Progressive encephalomyelitis with rigidity and myoclonus (PERM) - Limbic encephalitis |
| Anti-mGluR5 | - Limbic encephalitis often associated with Hodgkin lymphoma, termed Ophelia syndrome - Presents with memory deficits, seizures, and psychiatric changes |
| Anti-MOG | - Neuromyelitis optica (Optic neuritis and transverse demyelinating disease) - Acute disseminated encephalomyelitis |
| Anti-IgLON5 IgG1/4 | - Sleep disorder (REM sleep dysfunction and sleep apnea) - Brainstem signs with bulbar symptoms, speaking and swallowing difficulty - Movement disorder, chorea, dystonia, and parkinsonism gait disturbances - Cognitive syndrome and behavioral disorder |
NMDAR, N-methyl-D-aspartate receptor; LGI1, Leucine-rich glioma-inactivated 1; CASPR2, Contactin-associated protein-like 2; AMPA receptor, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; GABA, gamma-aminobutyric acid; DPPX, Dipeptidyl-peptidase-like protein-6; mGluR5, metabotropic glutamate receptor 5.
Clinical, diagnostic and therapeutic challenges and pitfalls in AIE
| Clinical challenges | Diagnostic pitfalls | Therapeutic challenges |
|---|---|---|
| 1. Lack of comprehensive clinical data; Insufficient data can hinder accurate diagnosis. 2. Diagnostic challenges for atypical cases; Unusual presentations may delay or complicate diagnosis. 3. Overlapping symptoms with other disorders; Symptoms may resemble other neurological conditions. 4. Variability in disease progression; The disease can manifest differently in different individuals. | 1. Inaccessible antibody testing; Results may take several weeks to obtain in many institutions. 2. Lack of autoantibodies does not exclude immune-mediated disorders; The absence of autoantibodies does not rule out an immune cause. 3. Positive autoantibody test does not guarantee accurate diagnosis; A positive test does not always mean the disorder is present. 4. Lack of specific biomarkers; Current biomarkers may not be definitive for all cases. | 1. Difficulty in early diagnosis; The initial symptoms can be nonspecific and may mimic other neurological or psychiatric diseases. 2. First-like vs. second-like therapies: While first-line therapies (steroids, IVIG, and plasmapheresis) can be effective for many, more aggressive second-line therapies, such as rituximab or cyclophosphamide is required in some patients. 3. Treatment resistance; some patients may not respond adequately to standard therapies. 4. Relapse and side effect; Even after successful initial treatment, AIE can relapse and potential side effects ranging from infections to organ toxicities. |
IVIG, intravascular immunoglobin treatment.
The diagnostic processes for seronegative AIE
| Diagnostic criteria | Description |
|---|---|
| Clinical presentation | Rapid onset of working memory deficits, altered mental status, or psychiatric symptoms. |
| MRI | Potential abnormalities, especially in the medial temporal lobes. |
| CSF analysis | Elevated white cell count (pleocytosis) or increased protein concentration. |
| EEG | Abnormalities such as epileptic or slow-wave activity. |
| Absence of alternate causes | Ruling out other potential causes like infections, metabolic disturbances, or tumors. |
| Response to immune therapy | Supportive of diagnosis if there's a response to treatments like steroids, IVIG, or plasmapheresis. |
| Autoantibody status | Absence of specific neural autoantibodies in serum. |
IVIG, intravascular immunoglobulin therapy.
References
- Abboud H, Probasco JC, Irani S, Ances B, Benavides DR, Bradshaw M, Christo PP, Dale RC, Fernandez-Fournier M, Flanagan EP, Gadoth A, George P, Grebenciucova E, Jammoul A, Lee ST, Li Y, Matiello M, Morse AM, Rae-Grant A, Rojas G, Rossman I, Schmitt S, Venkatesan A, Vernino S, Pittock SJ, Titulaer MJ; Autoimmune Encephalitis Alliance Clinicians Network (2021) Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management. J Neurol Neurosurg Psychiatry 92:757-768


- Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, Cortese I, Dale RC, Gelfand JM, Geschwind M, Glaser CA, Honnorat J, Höftberger R, Iizuka T, Irani SR, Lancaster E, Leypoldt F, Prüss H, Rae-Grant A, Reindl M, Rosenfeld MR, Rostásy K, Saiz A, Venkatesan A, Vincent A, Wandinger KP, Waters P, Dalmau J (2016) A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 15:391-404
- Vogrig A, Gigli GL, Nilo A, Pauletto G, Valente M (2022) Seizures, epilepsy, and NORSE secondary to autoimmune encephalitis: a practical guide for clinicians. Biomedicines 11:44
- Dalmau J, Tüzün E, Wu HY, Masjuan J, Rossi JE, Voloschin A, Baehring JM, Shimazaki H, Koide R, King D, Mason W, Sansing LH, Dichter MA, Rosenfeld MR, Lynch DR (2007) Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 61:25-36
- Brouwer B, Biemond A (1938) Les affections parenchymateuses du cervelet et leur signification au point de vue de l'anatomie et le physiologie de cet organe. J Belge Neurol Psychiatr 38:691-757
- Winer S, Astsaturov I, Cheung R, Gunaratnam L, Kubiak V, Cortez MA, Moscarello M, O'Connor PW, McKerlie C, Becker DJ, Dosch HM (2001) Type I diabetes and multiple sclerosis patients target islet plus central nervous system autoantigens; nonimmunized nonobese diabetic mice can develop autoimmune encephalitis. J Immunol 166:2831-2841
- Becher B, Durell BG, Noelle RJ (2002) Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest 110:493-497
- Najjar S, Pearlman D, Devinsky O, Najjar A, Nadkarni S, Butler T, Zagzag D (2013) Neuropsychiatric autoimmune encephalitis without VGKC-complex, NMDAR, and GAD autoantibodies: case report and literature review. Cogn Behav Neurol 26:36-49
- Maat P, de Beukelaar JW, Jansen C, Schuur M, van Duijn CM, van Coevorden MH, de Graaff E, Titulaer M, Rozemuller AJ, Sillevis Smitt P (2015) Pathologically confirmed autoimmune encephalitis in suspected Creutzfeldt-Jakob disease. Neurol Neuroimmunol Neuroinflamm 2:e178
- Dalmau J, Graus F (2023) Diagnostic criteria for autoimmune encephalitis: utility and pitfalls for antibody-negative disease. Lancet Neurol 22:529-540
- Lee WJ, Lee HS, Kim DY, Lee HS, Moon J, Park KI, Lee SK, Chu K, Lee ST (2022) Seronegative autoimmune encephalitis: clinical characteristics and factors associated with outcomes. Brain 145:3509-3521
- Simabukuro MM, Silva GDD, Castro LHM, Lucato LT (2022) A critical review and update on autoimmune encephalitis: understanding the alphabet soup. Arq Neuropsiquiatr 80(5 Suppl 1):143-158
- Patel A, Meng Y, Najjar A, Lado F, Najjar S (2022) Autoimmune encephalitis: a physician's guide to the clinical spectrum diagnosis and management. Brain Sci 12:1130
- Lucas SB, Wong KT, Nightingale S, Miller RF (2021) HIV-associated CD8 encephalitis: a UK case series and review of histopathologically confirmed cases. Front Neurol 12:628296
- Sharma R, Spradley T, Campbell M, Biyani S, Singhal P, Elkhider H, Nalleballe K, Gokden M, Kumar M, Kapoor N (2022) CD8 encephalitis: a diagnostic dilemma. Diagnostics (Basel) 12:2687
- Skrobot OA, Attems J, Esiri M, Hortobágyi T, Ironside JW, Kalaria RN, King A, Lammie GA, Mann D, Neal J, Ben-Shlomo Y, Kehoe PG, Love S (2016) Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain 139:2957-2969
- Esiri MM, Wilcock GK, Morris JH (1997) Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry 63:749-753
- Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, Gadoth A, Smith CY, Bryant SC, Klein CJ, Aksamit AJ, Toledano M, Boeve BF, Tillema JM, Flanagan EP (2018) Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol 83:166-177
- Venkatesan A, Tunkel AR, Bloch KC, Lauring AS, Sejvar J, Bitnun A, Stahl JP, Mailles A, Drebot M, Rupprecht CE, Yoder J, Cope JR, Wilson MR, Whitley RJ, Sullivan J, Granerod J, Jones C, Eastwood K, Ward KN, Durrheim DN, Solbrig MV, Guo-Dong L, Glaser CA; International Encephalitis Consortium (2013) Case definitions, diagnostic algorithms, and priorities in encephalitis: consensus statement of the international encephalitis consortium. Clin Infect Dis 57:1114-1128
- Uy CE, Binks S, Irani SR (2021) Autoimmune encephalitis: clinical spectrum and management. Pract Neurol 21:412-423
- Varley JA, Strippel C, Handel A, Irani SR (2023) Autoimmune encephalitis: recent clinical and biological advances. J Neurol 270:4118-4131
- Anurat K, Watcharakuldilok P, Sakpichaisakul K, Khongkhatithum C, Mahasirimongkol S, Kunhapan P, Inunchot W, Wattanapokayakit S, Munggaranonchai O, Thampratankul L (2022) HLA-DRB1*1502 is associated with anti-N-methyl-D-aspartate receptor encephalitis in Thai children. Pediatr Neurol 134:93-99
- Yan L, Zhang S, Huang X, Tang Y, Wu J (2021) Clinical study of autonomic dysfunction in patients with anti-NMDA receptor encephalitis. Front Neurol 12:609750
- Kim TJ, Lee ST, Moon J, Sunwoo JS, Byun JI, Lim JA, Shin YW, Jun JS, Lee HS, Lee WJ, Yang AR, Choi Y, Park KI, Jung KH, Jung KY, Kim M, Lee SK, Chu K (2017) Anti-LGI1 encephalitis is associated with unique HLA subtypes. Ann Neurol 81:183-192
- Schiff P, Muñiz-Castrillo S, Do LD, Fantini ML, Chanson E, Rogemond V, Honnorat J, Poncet-Megemont L (2023) Anti-LGI1 encephalitis with co-occurring IgLON5 antibodies: clinical features and human leukocyte antigen haplotypes. Neurol Neuroimmunol Neuroinflamm 10:e200126
- Binks S, Varley J, Lee W, Makuch M, Elliott K, Gelfand JM, Jacob S, Leite MI, Maddison P, Chen M, Geschwind MD, Grant E, Sen A, Waters P, McCormack M, Cavalleri GL, Barnardo M, Knight JC, Irani SR (2018) Distinct HLA associations of LGI1 and CASPR2-antibody diseases. Brain 141:2263-2271
- da Rocha AJ, Nunes RH, Maia AC Jr, do Amaral LL (2015) Recognizing autoimmune-mediated encephalitis in the differential diagnosis of limbic disorders. AJNR Am J Neuroradiol 36:2196-2205
- Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, Friedman D, Skeen MB, Grisold W, Kimura A, Ohta K, Iizuka T, Guzman M, Graus F, Moss SJ, Balice-Gordon R, Dalmau J (2010) Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 9:67-76
- Wesselingh R, Butzkueven H, Buzzard K, Tarlinton D, O'Brien TJ, Monif M (2019) Innate immunity in the central nervous system: a missing piece of the autoimmune encephalitis puzzle?. Front Immunol 10:2066
- Abu-Abaa M, Chadalawada S, Jumaah O, Abubakar M, Landau D (2023) Important clues for the diagnosis of anti-LGI1-antibody autoimmune encephalitis: a case report. Cureus 15:e34222
- Salama S, Khan M, Pardo S, Izbudak I, Levy M (2019) MOG antibody-associated encephalomyelitis/encephalitis. Mult Scler 25:1427-1433
- Chung HY, Wickel J, Voss A, Ceanga M, Sell J, Witte OW, Geis C (2019) Autoimmune encephalitis with anti-IgLON5 and anti-GABAB-receptor antibodies: a case report. Medicine (Baltimore) 98:e15706
- Madetko N, Marzec W, Kowalska A, Przewodowska D, Alster P, Koziorowski D (2022) Anti-IgLON5 disease - the current state of knowledge and further perspectives. Front Immunol 13:852215
- Dai X, Kuang L, Feng L, Yi X, Tang W, Liao Q, Long X, Wang J, Li J, Yang H, Xiao B, Li G, Chen S (2020) Anti-dopamine receptor 2 antibody-positive encephalitis in adolescent. Front Neurol 11:471
- Bauer J, Bien CG (2016) Neuropathology of autoimmune encephalitides. Handb Clin Neurol 133:107-120
- Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, Jellinger KA, Reuss DE, Ribalta T, Schlegel J, Sutton I, Lassmann H, Bauer J (2012) Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 135(Pt 5):1622-1638
- Dalmau J, Graus F (2018) Antibody-mediated encephalitis. N Engl J Med 378:840-851
- Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, Gleichman AJ, Balice-Gordon R, Rosenfeld MR, Lynch D, Graus F, Dalmau J (2014) Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 13:167-177
- Beckhove P, Warta R, Lemke B, Stoycheva D, Momburg F, Schnölzer M, Warnken U, Schmitz-Winnenthal H, Ahmadi R, Dyckhoff G, Bucur M, Jünger S, Schueler T, Lennerz V, Woelfel T, Unterberg A, Herold-Mende C (2010) Rapid T cell-based identification of human tumor tissue antigens by automated two-dimensional protein fractionation. J Clin Invest 120:2230-2242
- Lee SK, Lee ST (2016) The laboratory diagnosis of autoimmune encephalitis. J Epilepsy Res 6:45-50
- van Steenhoven RW, Titulaer MJ (2022) Seronegative autoimmune encephalitis: exploring the unknown. Brain 145:3339-3340
- Dubey D, Wilson MR, Clarkson B, Giannini C, Gandhi M, Cheville J, Lennon VA, Eggers S, Devine MF, Mandel-Brehm C, Kryzer T, Hinson SR, Khazaie K, Hales C, Kattah J, Pavelko KD, Andrews P, Eaton JE, Jitprapaikulsan J, Mills JR, Flanagan EP, Zekeridou A, Leibovich B, Fryer J, Torre M, Kaufman C, Thoreson JB, Sagen J, Linnoila JJ, DeRisi JL, Howe CL, McKeon A, Pittock SJ (2020) Expanded clinical phenotype, oncological associations, and immunopathologic insights of paraneoplastic Kelch-like protein-11 encephalitis. JAMA Neurol 77:1420-1429
- Terziroli Beretta-Piccoli B, Mieli-Vergani G, Vergani D (2022) Autoimmmune hepatitis. Cell Mol Immunol 19:158-176
- Santana LM, Valadares EA, Ferreira-Júnior CU, Santos MF, Albergaria BH, Rosa-Júnior M (2020) CD8 + T-lymphocyte encephalitis: a systematic review. AIDS Rev 22:112-122
- Collier JL, Weiss SA, Pauken KE, Sen DR, Sharpe AH (2021) Not-so-opposite ends of the spectrum: CD8+ T cell dysfunction across chronic infection, cancer and autoimmunity. Nat Immunol 22:809-819
- Nabizadeh F, Balabandian M, Sodeifian F, Rezaei N, Rostami MR, Naser Moghadasi A (2022) Autoimmune encephalitis associated with COVID-19: a systematic review. Mult Scler Relat Disord 62:103795
- Mosaad YM (2015) Clinical role of human leukocyte antigen in health and disease. Scand J Immunol 82:283-306
- Cruz-Tapias P, Castiblanco J, Anaya JM (2013) HLA and proteasome expression body map. BMC Med Genomics 11:36
- Boegel S, Löwer M, Bukur T, Sorn P, Castle JC, Sahin U (2018) HLA and proteasome expression body map. BMC Med Genomics 11:36
- Wang ZX, Wan Q, Xing A (2020) HLA in Alzheimer's disease: genetic association and possible pathogenic roles. Neuromolecular Med 22:464-473
- Hobson BD, Sulzer D (2022) Neuronal presentation of antigen and its possible role in Parkinson's disease. J Parkinsons Dis 12(s1):S137-S147
- Ulvestad E, Williams K, Bø L, Trapp B, Antel J, Mørk S (1994) HLA class II molecules (HLA-DR, -DP, -DQ) on cells in the human CNS studied in situ and in vitro. Immunology 82:535-541
- Couture A, Garnier A, Docagne F, Boyer O, Vivien D, Le-Mauff B, Latouche JB, Toutirais O (2019) HLA-class II artificial antigen presenting cells in CD4+ T cell-based immunotherapy. Front Immunol 10:1081
- Naito T, Okada Y (2022) HLA imputation and its application to genetic and molecular fine-mapping of the MHC region in autoimmune diseases. Semin Immunopathol 44:15-28
- Sabbatino F, Liguori L, Polcaro G, Salvato I, Caramori G, Salzano FA, Casolaro V, Stellato C, Col JD, Pepe S (2020) Role of human leukocyte antigen system as a predictive biomarker for checkpoint-based immunotherapy in cancer patients. Int J Mol Sci 21:7295
- Peris Sempere V, Muñiz-Castrillo S, Ambati A, Binks S, Pinto AL, Rogemond V, Pittock SJ, Dubey D, Geschwind MD, Gelfand JM, Dilwali S, Lee ST, Knight J, Elliott KS, Irani S, Honnorat J, Mignot E (2022) Human leukocyte antigen association study reveals DRB1*04:02 effects additional to DRB1*07:01 in anti-LGI1 encephalitis. Neurol Neuroimmunol Neuroinflamm 9:e1140
- Shepshelovich D, Leibovitch C, Klein A, Zoldan S, Milo G, Shochat T, Rozen-zvi B, Gafter-Gvili A, Lahav M (2015) The syndrome of inappropriate antidiuretic hormone secretion: distribution and characterization according to etiologies. Eur J Intern Med 26:819-824
- Ayus JC, Krothapalli RK, Arieff AI (1987) Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med 317:1190-1195
- Mori D, Nagayama I, Yamaguchi Y, Itano S, Imakita N, Takeji M, Yamauchi A (2013) Hyponatremia associated with demyelinating disease of the nervous system. CEN Case Rep 2:84-89