Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2024; 33(1): 36-45
Published online February 29, 2024
https://doi.org/10.5607/en23023
© The Korean Society for Brain and Neural Sciences
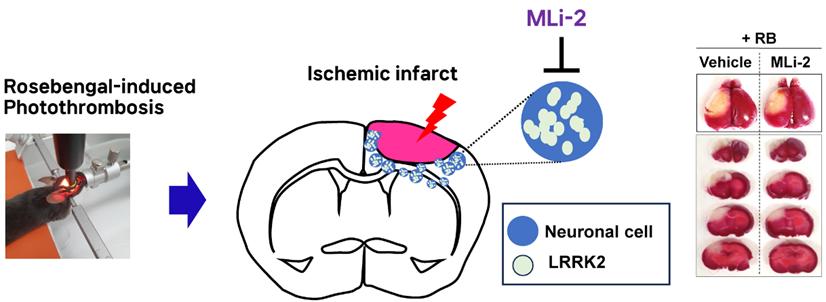
Pharmacological Inhibition of LRRK2 Exhibits Neuroprotective Activity in Mouse Photothrombotic Stroke Model
Jeong-Ah Hwang1, Seung Kyu Choi2,3, Seong Hwan Kim1* and Dong Woon Kim2,3,4*
1Center for Rare Disease Therapeutic Technology, Therapeutic & Biotechnology Division, Korea Research Institute of Chemical Technology, Daejeon 34114,
2Department of Medical Science, Chungnam National University College of Medicine, Daejeon 35015,
3Department of Anatomy and Cell Biology, Brain Research Institute, Chungnam National University College of Medicine, Daejeon 35015,
4Department of Anatomy & Developmental Biology, Kyung Hee University School of Dentistry, Seoul 02447, Korea
Correspondence to: *To whom correspondence should be addressed.
Seong Hwan Kim, TEL: 82-42-860-7687, FAX: 82-42-861-4246
e-mail: hwan@krict.re.kr
Dong Woon Kim, TEL: 82-2-961-9302, FAX: 82-2-961-9594
e-mail: visnu528@cnu.ac.kr, visnu528@khu.ac.kr
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Leucine-rich repeat kinase 2 (LRRK2) mutations are the most common cause of Parkinson’s disease (PD). Interestingly, recent studies have reported an increased risk of stroke in patients with PD harboring LRRK2 mutations, but there is no evidence showing the functional involvement of LRRK2 in stroke. Here, we found that LRRK2 kinase activity was significantly induced in the Rose-Bengal (RB) photothrombosis-induced stroke mouse model. Interestingly, stroke infarct volumes were significantly reduced, and neurological deficits were diminished by pharmacological inhibition of LRRK2 kinase activity using MLi-2, a brain-penetrant LRRK2 kinase inhibitor. Immunohistochemical analysis showed p-LRRK2 level in stroke lesions, co-localizing with mitophagy-related proteins (PINK, Parkin, LC3B, cytochrome c), suggesting their involvement in stroke progression. Overlapping p-LRRK2 with cytochrome c/TUNEL/JC-1 (an indicator of mitochondrial membrane potential) puncta in RB photothrombosis indicated LRRK2-induced mitochondrial apoptosis, which was blocked by MLi-2. These results suggest that pharmacological inhibition of LRRK2 kinase activity could attenuate mitochondrial apoptosis, ultimately leading to neuroprotective potential in stroke progression. In conclusion, LRRK2 kinase activity might be neuro-pathogenic due to impaired mitophagy in stroke progression, and pharmacological inhibition of LRRK2 kinase activity could be beneficial in reducing the risk of stroke in patients with LRRK2 mutations.
Graphical Abstract
Keywords: Ischemic stroke, Photothrombosis, LRRK2, Mitochondria, Apoptosis
INTRODUCTION
Leucine-rich repeat kinase 2 (LRRK2) mutations are recognized as a major contributing factor to Parkinson’s disease (PD) [1-3]. In fact, both familial and sporadic forms of PD share common pathological mechanisms associated with LRRK2 mutations, whether through genetic variations or non-coding alterations at its locus [4-7]. Pathogenic mutations in the LRRK2 gene have been demonstrated to enhance its kinase activity, and this abnormal increase in LRRK2 kinase activity is believed to play a role in the pathogenesis of PD [8-12]. Consequently, pharmacological inhibition of LRRK2 kinase activity has been explored as a potential therapeutic strategy for PD patients.
PD primarily manifests as the loss of dopaminergic neurons in the brain, leading to motor symptoms such as tremors, rigidity, and bradykinesia. However, PD is not limited to motor symptoms; it also affects the autonomic nervous system, which controls involuntary functions like heart rate, blood pressure, and digestion. The interplay between impaired sympathetic vasoconstriction and compensatory autonomic responses may disrupt blood flow regulation and increase the risk of stroke in PD patients. For instance, neurogenic orthostatic hypotension can result in reduced cerebral blood flow, rendering the brain more susceptible to ischemic events [13, 14].
Notably, recent studies have reported an elevated risk of stroke among PD patients [15, 16]. Specifically, individuals with PD attributed to mutations in the LRRK2 gene appear to have a higher stroke risk than those with PD caused by other factors. Moreover, when compared to PD patients with mutations in other genes such as parkin and glucocerebrosidase, who exhibit stroke risk levels similar to controls, it suggests that LRRK2-associated mechanisms may significantly contribute to the heightened stroke risk in PD patients with LRRK2 mutations. Hence, we posit that pharmacological inhibition of LRRK2 kinase activity may offer potential benefits in reducing the risk of stroke. To test this hypothesis, we conducted an investigation into the functional role of LRRK2 and the neuroprotective effects of an LRRK2 inhibitor in a mouse stroke model.
MATERIALS AND METHODS
Rose Bengal (RB) photothrombosis mice model
Male C57BL/6 mice (7~8 weeks old) were obtained from Damul Science (Daejeon, Korea). To induce photothrombosis, a photosensitive dye, Rose Bengal (RB), dissolved in PBS, was injected through the tail vein at a dosage of 0.5 mg/100 μl. To aggregate the dye, an area of 0.6 mm in diameter in the right cerebral cortex was locally illuminated with green light (535±25 nm) using an LED lamp (ZEISS, CL 6000 LED) for 3 minutes. For pharmacological inhibition of LRRK2, starting from 2 days before RB photothrombosis modeling, 30 mg/kg of MLi-2 (Selleck Chemicals, TX; n=10) or the vehicle control (n=10) were administered via oral gavage once daily. Mice were euthanized 72 hours after modeling.
Immunohistochemistry
C57BL/6 male mice were anesthetized with isoprene + 1% O2 and perfused intracardially with 4% paraformaldehyde in PBS buffer. To prepare cryostat sections, post-fixed brains were cryoprotected in 30% sucrose (wt/vol) in PBS for 48 hours at 4°C, embedded in Tissue-Tek OCT compound (Sakura Finetek USA, CA), and frozen on dry ice. Cryostat sections were blocked for 1 hour at room temperature using a solution containing 5% normal goat serum (Vector Laboratories Inc., CA) in 0.01 M PBS with 0.5% Triton X-100. Subsequently, sections were incubated with primary antibodies diluted in blocking solution at 4°C overnight. The following primary antibodies were used: anti-LRRK2 (Abcam, MA), anti-p-LRRK2 (Ser1292) (Abcam), anti-NeuN (Millipore, MO), anti-GFAP (Dako, Glostrup Municipality, Denmark), anti-PINK1 (Novus Biologicals, CO), anti-Parkin (Abcam), anti-LC3B (Cell signaling, MA), anti-cytochrome C (Abcam), and JC-1 (Invitrogen, MA) dye. Sections were then incubated with Cy3- and Cy2-conjugated secondary antibodies (Jackson ImmunoResearch, PA) for 2 hours at room temperature. Following PBS washes, tissues were incubated with DAPI for 3 minutes. After three washes, tissues were mounted on slides and visualized under a fluorescence microscope. The Click-iTTM TUNEL Alexa FluorTM 488 Imaging Assay (Thermo Fisher Scientific, MA) was used to analyze neuronal apoptosis following the manufacturer’s instructions.
Infarct volume quantification
Brains were removed 24 hours after RB photothrombosis, sectioned into 2 mm slices, and immersed in 1% TTC (Sigma-Aldrich, MO) solution at 37°C for 15 minutes, followed by fixation with 4% paraformaldehyde. ImageJ software was utilized to measure the areas of ischemic damage for each brain slice. Lack of TTC staining defined the core, while red staining indicated viable tissue. The total infarction volume was calculated as the sum of core and penumbra volumes. Damage volume was expressed as a percentage of the ipsilateral hemisphere volume (damage/total volume × 100). This calculation was performed for 4 hemispheric brain slices per animal.
Neurological deficits measurement
The neurological status of the experimental animals was assessed using an adapted five-point scale test based on Yang et al [17]. The criteria were as follows: Grade 0=normal neurological function, Grade 1=failure to extend the right forepaw, Grade 2=circling, Grade 3=leaning to the contralateral side at rest, Grade 4=no spontaneous motor activity, Grade 5=dead. Neurological deficit was evaluated for 24 hours after RB photothrombosis. Motor performance was assessed using the Wire Hang test, conducted 1 day and 3 days after RB photothrombosis. A wire cage top (18 inches×9 inches) with taped edges was employed for this experiment. Mice were placed on the metallic wire, the top was inverted, and latency to fall was recorded. This was repeated three times with a 5-minute interval between attempts. The cutoff time was set at 60 seconds, and the mean latency to fall was determined [18].
Image analysis
The puncta measurement data were analyzed with scripts written in ImageJ. The analyses were performed using a script written in ImageJ for batch processing to handle the hundreds of ROIs imaged per slide.
Statistical analysis
All data were presented as mean±SEM. Statistical differences among more than two groups were analyzed using one-way ANOVA followed by Tukey post hoc analysis. Differences between two groups were analyzed using a t-test, and a p-value<0.05 was considered statistically significant. Statistical calculations were performed using the GraphPad Prism program.
RESULTS
Induction of LRRK2 and p-LRRK2 in the Rose Bengal Photothrombotic Stroke Model
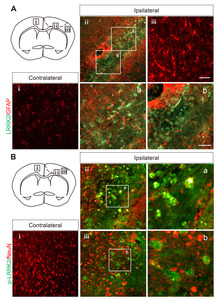
The functional involvement of LRRK2 in the risk of stroke was elucidated by its expression and activation in the Rose Bengal (RB) photothrombosis model, which simulates human stroke induced by blood clots [19, 20]. Histological analysis of cortex lesions 3 days after RB photothrombosis revealed an increased number of cells with LRRK2-positive puncta in infarct lesions, suggesting that the induction of LRRK2 protein expression might be associated with the risk of stroke (Fig. 1). Notably, LRRK2 puncta were observed in the penumbra (the boundary of the infarct), but not in the infarct core lesions. This is in line with previous studies indicating that endogenous LRRK2 is present at low levels in most cell types [21], LRRK2 expression was not observed in the contralateral areas. Similarly, p-LRRK2 (Ser1292), which represents LRRK2 kinase activity, was highly detected in the in the penumbra (the boundary of the infarct) but absent in the contralateral areas.
Pharmacological inhibition of LRRK2 kinase activity mitigates neurological deficits in the RB photothrombosis model
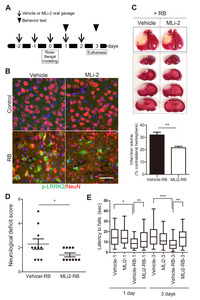
The hypothesis that LRRK2 activation under stroke conditions could exacerbate brain damage suggested the therapeutic potential of LRRK2 inhibition in treating stroke. To investigate this, we utilized MLi-2, a highly selective LRRK2 inhibitor capable of penetrating the blood-brain barrier [22]. Mice received 30 mg/kg of MLi-2 daily via oral gavage, starting 2 days before RB modeling and continuing for 2 days after modeling (Fig. 2A). The repeated administration of 30 mg/kg of MLi-2 in mice markedly inhibited p-LRRK2 (Ser1292) in the penumbra and exhibited the excellent tolerability and no adverse effects (Fig. 2B). In contrast, the control groups treated with the vehicle did not exhibit any LRRK2 activation. As seen in Fig. 1 and 2B, RB modeling strongly activated LRRK2 in the penumbra (the boundary of the infarct), while MLi-2 administration attenuated this RB-induced LRRK2 activation. Compared to the RB-only groups, the MLi-2-treated RB group displayed weaker p-LRRK2 signals that did not form puncta, suggesting that MLi-2 treatment effectively reduces LRRK2 kinase activity in the context of RB-induced stroke.
Importantly, the MLi-2 treatment group exhibited a one-third lower infarct volume than the vehicle treatment group following RB photothrombosis (infarct volume in the vehicle-treated RB groups, 32.00%±2.21%; in the MLi-2-treated RB groups, 21.60%±1.21%; p=0.003). Additionally, analysis of neurological deficit scores (vehicle-treated RB groups, 2.30±0.42; MLi-2-treated RB groups, 1.40±0.16; p=0.031) and results from the wire hang behavior test, conducted 3 days after RB photothrombosis (vehicle-treated RB groups, 8.23±1.01; MLi-2-treated RB groups, 14.77±1.65; p=0.0013), indicated that MLi-2 treatment significantly attenuated RB photothrombosis-induced neurological deficits (Fig. 2D, E).
p-LRRK2 co-localization with mitochondrial apoptosis signaling proteins
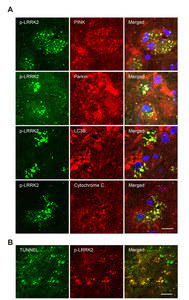
The punctate LRRK2 expression in the RB photothrombosis model suggested a potential connection to the autophagy process, which involves the delivery of intracellular components to the lysosome for degradation [23, 24]. One well-studied form of autophagy is macro-autophagy, and the degradation of mitochondria via recruitment to autophagosomes by the autophagy mediator, LC3, is known as mitophagy. Damaged mitochondria can trigger apoptotic stress and cell death through the release of cytochrome c. Importantly, LRRK2 mutations have been shown to impair PINK/Parkin-dependent mitophagy in several studies [25-28]. To investigate whether LRRK2 activation in the RB photothrombosis model could be related to PINK/Parkin-dependent mitophagy, we analyzed the co-localization of p-LRRK2 (Ser1292) with mitophagy-related proteins (PINK1, Parkin, LC3B) [29]. Additionally, we examined its co-localization with cytochrome c, a marker of mitochondrial apoptosis, as several studies have reported that LRRK2 mutations could induce cell toxicity via mitochondria-dependent apoptosis [23, 30]. As shown in Fig. 3A, p-LRRK2 (Ser1292) strongly co-localized with PINK1, Parkin, LC3B, and cytochrome c in cortical infarct penumbra at 72 hours post-stroke. Furthermore, late-stage apoptosis was assessed by detecting DNA & mitochondrial fragmentation using the TUNEL assay in core infarcts at 72 hours post-stroke (Fig. 3B), revealing an overlap between p-LRRK2-positive puncta and TUNEL-positive puncta.
Pharmacological inhibition of LRRK2 attenuated mitochondrial apoptosis
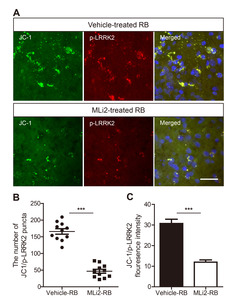
A distinctive feature of the early stages of apoptosis is the disruption of active mitochondria, including changes in mitochondrial membrane potentials. The JC-1 dye, capable of intercalating into the mitochondrial membrane, is commonly used to evaluate mitochondrial membrane potential changes in apoptosis [31, 32]. To elucidate the relationship between LRRK2 activation and mitochondrial apoptosis, brain tissues obtained from the Rose Bengal photothrombosis model were stained with JC-1. Consistent with Fig. 3, p-LRRK2 puncta were found to co-localize with JC-1-positive mitochondrial puncta, suggesting that elevated p-LRRK2 could be associated with mitochondrial apoptosis (Fig. 4A). Furthermore, the number of JC-1 and p-LRRK2-positive puncta was significantly lower in the MLi-2-treated RB group compared to the vehicle-treated RB group (Fig. 4A, B). This observation was in line with the results showing JC-1 and p-LRRK2-positive fluorescence intensity (Fig. 4C).
DISCUSSION
This study represents the first report of the possible involvement of LRRK2 expression and its activation in an in vivo stroke mouse model. RB photothrombosis induced the expressions of both LRRK2 and p-LRRK2 (Ser1292), which has been demonstrated as a physiological and direct marker of LRRK2 kinase activity [5, 6]. Moreover, LRRK2 and p-LRRK2-positive puncta were observed in the infarct boundary but not in infarct core lesions, suggesting that the induction/activation of LRRK2 in the progression of stroke may determine the cell's fate to die, as the functional abnormality of LRRK2 has been known to be pathogenic [10-12].
Interestingly, pharmacological inhibition of LRRK2 using MLi-2 demonstrated an alleviation of stroke-induced neurological deficits in the in vivo stroke mouse model. The MLi-2-treated RB group showed significant improvements in neurological deficit scores and behavioral test results. Considering the higher stroke risk in patients with PD with LRRK2 mutations [15] and the reports indicating that LRRK2 mutations induce an abnormal increase in its kinase activity contributing to PD pathology, our results suggest that LRRK2 inhibition in PD treatment could reduce the risk of stroke in patients with PD as well as slow the progression of PD.
So, how does pharmacological inhibition of LRRK2 work in stroke? The pattern of LRRK2/p-LRRK2-positive puncta suggests a link to physiological vesicle-mediated signaling pathways, such as autophagy. Increased autophagosomes and autolysosomes are often seen in degenerating or dying neurons, representing autophagic stress [33, 34]. These phenotypes, including autophagic vacuoles, have been observed in various neurodegenerative diseases, including PD [35-38]. Additionally, the functional abnormality of LRRK2 caused by mutations has been shown to result in mitochondrial dysfunction and impaired autophagy of mitochondria, known as mitophagy [25-28]. Mitophagy has strong ties to PD, as it was discovered that PINK1 and Parkin, two other genes mutated in familial PD, initiate mitophagy through mitochondrial depolarization [39-42]. Interestingly, several studies have reported that pathogenic LRRK2 mutants impair mitophagy by reducing mitochondrial trafficking [25, 26] and disrupting the recruitment of mitochondrial proteins like Drp1 and Parkin [27] and inhibited small GTPase RAB10 interaction with the autophagy receptor OPTN [26]. Consequently, impaired mitophagy has been proposed as a common factor in PD. The activation of LRRK2 in PINK/Parkin-dependent mitophagy in RB photothrombosis suggests that the LRRK2-driven PINK/Parkin-dependent mitophagy mechanisms observed in PD may also be involved in the progression of stroke.
Bae et al. [43], group found that various in vitro and in vivo models of traumatic brain injury (TBI) markedly enhanced LRRK2 expression in neurons and also increased the level of hypoxia-inducible factor (HIF)-1α. They also revealed direct binding of HIF-1α in LRRK2 proximal promoter and found that HIF-1α-dependent transcriptional induction of LRRK2 exacerbated neuronal cell death following injury. These studies are very similar to our findings, and the mechanism of HIF-1a dependent transcriptional induction of LRRK2 may also operate in stroke, and future research is needed. In addition, the co-localization of p-LRRK2-positive puncta with cytochrome c/TUNEL/JC-1-positive puncta in RB photothrombosis suggests that elevated p-LRRK2 may induce mitochondrial apoptosis in infarct lesions. Importantly, the number of JC-1 and p-LRRK2-positive puncta in RB photothrombosis was significantly reduced by MLi-2 treatment, indicating that pharmacological inhibition of LRRK2 can attenuate mitochondrial apoptosis and ultimately exhibit neuroprotective potential in the progression of stroke.
In summary, LRRK2 activation may contribute to neuro-pathogenesis through impaired mitophagy in the progression of stroke, and pharmacological inhibition of LRRK2 may hold promise in reducing the risk of stroke and delaying the progression of PD in patients with LRRK2 mutations.
ACKNOWLEDGEMENTS
This work was supported by the project grants (SI2131-10, SI2231-10 & KK2331-10 funded by KRICT), the National Research Foundation of Korea (NRF-2022R1A2B5B02001886), and by research fund of Chungnam National University.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ETHICAL APPROVAL
All protocols performed in accordance with the Chungnam National University hospital Guide for the Care and Use of Laboratory Animals and were approved by the University of Chungnam Institutional Animal Care and Use Committee.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F (2002) A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 51:296-301

- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Martí Carrera I, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44:595-600
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 4:601-607
- Aasly JO, Toft M, Fernandez-Mata I, Kachergus J, Hulihan M, White LR, Farrer M (2005) Clinical features of LRRK2-associated Parkinson's disease in central Norway. Ann Neurol 57:762-765
- Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ, Toft M (2005) Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 76:672-680

- Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, Zharikov A, Van Laar A, Stepan AF, Lanz TA, Kofler JK, Burton EA, Alessi DR, Hastings TG, Greenamyre JT (2018) LRRK2 activation in idiopathic Parkinson's disease. Sci Transl Med 10:eaar5429
- Fraser KB, Rawlins AB, Clark RG, Alcalay RN, Standaert DG, Liu N (2016) Ser(P)-1292 LRRK2 in urinary exosomes is elevated in idiopathic Parkinson's disease. Mov Disord 31:1543-1550
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 102:16842-16847
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M (2006) The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet 15:223-232
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 23:329-341
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9:1231-1233
- Pischedda F, Cirnaru MD, Ponzoni L, Sandre M, Biosa A, Carrion MP, Marin O, Morari M, Pan L, Greggio E, Bandopadhyay R, Sala M, Piccoli G (2021) LRRK2 G2019S kinase activity triggers neurotoxic NSF aggregation. Brain 144:1509-1525
- Coon EA, Cutsforth-Gregory JK, Benarroch EE (2018) Neuropathology of autonomic dysfunction in synucleinopathies. Mov Disord 33:349-358
- Suri JS, Paul S, Maindarkar MA, Puvvula A, Saxena S, Saba L, Turk M, Laird JR, Khanna NN, Viskovic K, Singh IM, Kalra M, Krishnan PR, Johri A, Paraskevas KI (2022) Cardiovascular/stroke risk stratification in Parkinson's disease patients using atherosclerosis pathway and artificial intelligence paradigm: a systematic review. Metabolites 12:312
- Elfil M, Bayoumi A, Sayed A, Aladawi M, Aboutaleb PE, Grieb L, Tolba H, Tinaz S (2023) Stroke in Parkinson's disease: a review of epidemiological studies and potential pathophysiological mechanisms. Acta Neurol Belg 123:773-783
- Macías-García D, Periñán MT, Muñoz-Delgado L, Jesús S, Jimenez-Jaraba MV, Buiza-Rueda D, Bonilla-Toribio M, Adarmes-Gómez A, Carrillo F, Gómez-Garre P, Mir P (2022) Increased stroke risk in patients with Parkinson's disease with LRRK2 mutations. Mov Disord 37:225-227
- Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H (1994) Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke 25:165-170
- Hwang JA, Shin N, Shin HJ, Yin Y, Kwon HH, Park H, Shin J, Kim SI, Kim DW, Song HJ (2021) Protective effects of ShcA protein silencing for photothrombotic cerebral infarction. Transl Stroke Res 12:866-878
- Talley Watts L, Zheng W, Garling RJ, Frohlich VC, Lechleiter JD (2015) Rose bengal photothrombosis by confocal optical imaging in vivo: a model of single vessel stroke. J Vis Exp 100:e52794
- Carmichael ST (2005) Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx 2:396-409
- Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, Dawson TM, West AB (2007) Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci 8:102
- Kluss JH, Mazza MC, Li Y, Manzoni C, Lewis PA, Cookson MR, Mamais A (2021) Preclinical modeling of chronic inhibition of the Parkinson's disease associated kinase LRRK2 reveals altered function of the endolysosomal system in vivo. Mol Neurodegener 16:17
- Iaccarino C, Crosio C, Vitale C, Sanna G, Carrì MT, Barone P (2007) Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum Mol Genet 16:1319-1326
- Yu L, Chen Y, Tooze SA (2018) Autophagy pathway: cellular and molecular mechanisms. Autophagy 14:207-215
- Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, Schüle B, Krainc D, Palmer TD, Wang X (2016) Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson's disease. Cell Stem Cell 19:709-724
- Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, Miller CC, Whitworth AJ, De Vos KJ (2014) Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat Commun 5:5245
- Bonello F, Hassoun SM, Mouton-Liger F, Shin YS, Muscat A, Tesson C, Lesage S, Beart PM, Brice A, Krupp J, Corvol JC, Corti O (2019) LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson's disease. Hum Mol Genet 28:1645-1660
- Wauters F, Cornelissen T, Imberechts D, Martin S, Koentjoro B, Sue C, Vangheluwe P, Vandenberghe W (2020)
LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16:203-222
- Kawajiri S, Saiki S, Sato S, Sato F, Hatano T, Eguchi H, Hattori N (2010) PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett 584:1073-1079
- Gonzalez-Hunt CP, Thacker EA, Toste CM, Boularand S, Deprets S, Dubois L, Sanders LH (2020) Mitochondrial DNA damage as a potential biomarker of LRRK2 kinase activity in LRRK2 Parkinson's disease. Sci Rep 10:17293
- Reers M, Smith TW, Chen LB (1991) J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 30:4480-4486
- Shibata M, Yamasaki N, Miyakawa T, Kalaria RN, Fujita Y, Ohtani R, Ihara M, Takahashi R, Tomimoto H (2007) Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 38:2826-2832
- Clarke PG (1990) Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 181:195-213
- Chu CT (2006) Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol 65:423-432
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol 12:25-31
- Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT (2003) Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol 13:473-481
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64:113-122
- Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M (1997) Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol 42:604-612
- Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795-803
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510:162-166
- Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85:257-273
- Yamano K, Wang C, Sarraf SA, Münch C, Kikuchi R, Noda NN, Hizukuri Y, Kanemaki MT, Harper W, Tanaka K, Matsuda N, Youle RJ (2018) Endosomal Rab cycles regulate Parkin-mediated mitophagy. Elife 7:e31326
- Bae YH, Joo H, Bae J, Hyeon SJ, Her S, Ko E, Choi HG, Ryu H, Hur EM, Bu Y, Lee BD (2018) Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell Death Dis 9:1125